When a generic drug hits the market, it’s not set in stone. Even small changes to how it’s made can trigger a full FDA re-evaluation. Many assume that once a generic is approved, it’s approved for life. But that’s not true. The FDA requires manufacturers to report and get approval for nearly every change to the manufacturing process, equipment, location, or ingredients - even if the final pill looks and works the same. Why? Because the law demands that every batch of a generic drug must be identical in safety, strength, purity, and quality to the brand-name version it copies. And if you change how you make it, you have to prove you didn’t change what it does.
What Kind of Changes Trigger FDA Review?
Not every tweak needs a full review. The FDA classifies manufacturing changes into three buckets: Prior Approval Supplements (PAS), Changes Being Effected (CBE), and Annual Reports (AR). Only PAS changes require the FDA to say yes before you can make the switch. These are the big ones - the kind that could affect how the drug behaves in your body.PAS triggers include:
- Switching to a new factory or moving production to a different country
- Changing the synthesis route for the active ingredient (how the drug molecule is built)
- Scaling up production by more than 50% in batch size
- Replacing key equipment like tablet presses or fillers with different models
- Introducing new excipients (inactive ingredients) not in the original approved formula
- Changing the drug’s release profile - for example, turning a quick-release tablet into extended-release
For example, if a company makes a generic blood pressure pill and decides to use a new type of coating to improve shelf life, that’s a PAS. Even if the coating is chemically safe, the FDA needs to confirm it doesn’t slow down how fast the drug gets into the bloodstream. If it does, the pill might not work as well. That’s why bioequivalence studies - tests proving the generic acts the same in the body as the brand - often become part of the submission.
Why Does the FDA Care So Much?
The core idea behind generic drugs is simple: same medicine, lower price. But that only works if the medicine is truly the same. The FDA doesn’t re-test safety or effectiveness for generics - they rely on the brand-name drug’s data. What they do test is whether the generic matches the original in how it’s made and how it behaves in the body. That’s called Chemistry, Manufacturing, and Controls (CMC). If you alter the manufacturing process, you risk altering the drug’s behavior - even slightly.Take a peptide drug, like insulin or a hormone treatment. These are complex molecules. A tiny impurity - say, 0.6% instead of the approved 0.5% - could trigger an immune reaction. The FDA requires manufacturers to prove every new impurity is safe. For these drugs, even a minor change in the fermentation tank or purification filter can mean a full PAS submission.
It’s not just about safety. Quality matters too. If a tablet crumbles during shipping because of a new binder, patients won’t get the right dose. If the drug degrades faster because of a new packaging material, it might expire too soon. The FDA’s job is to make sure every pill, capsule, or injection works exactly as intended - no matter where or when it was made.
How Long Does Re-Evaluation Take?
A PAS can take anywhere from 8 to 18 months to get approved. The average is about 10 months. That’s a long time for a company that’s trying to fix a production problem or improve efficiency. For comparison, a CBE-30 (a change you can make while waiting for FDA feedback) takes about 3 months to review. But you can’t use CBE-30 for major changes.Why so slow? Because the FDA needs to review hundreds of pages of data: stability studies, analytical methods, validation reports, facility inspections, and sometimes clinical bioequivalence data. In 2022, over two-thirds of PAS submissions needed at least one “complete response letter” - meaning the FDA asked for more information. Common reasons? Unclear analytical methods, missing stability data, or insufficient comparison between old and new processes.
One real case: a manufacturer increased batch size by 30% for a common cholesterol pill. They spent six months running stability tests, validating the new equipment, and documenting every step. Then they submitted the PAS. It took 14 months for approval. During that time, they couldn’t sell the new version. That’s a huge financial hit for a low-margin product.

Why Do Companies Avoid Making Improvements?
Here’s the catch: making a better process often costs more than it saves. Submitting a PAS can cost $287,500 on average - not counting internal labor, testing, or lost sales during review. For a generic drug that sells for pennies per pill, that’s a hard sell to shareholders.Many manufacturers, especially smaller ones, avoid upgrades entirely. One quality manager on Reddit described how their company shelved a tablet press upgrade because the FDA review would take 18 months - and they didn’t know if it would even be approved. They kept using the old machine, even though it broke down more often. It was cheaper to deal with downtime than regulatory risk.
There’s also inconsistency. One FDA reviewer might accept a change that another rejects. A 2023 survey found 65% of manufacturers complained about unpredictable feedback. A change that got approved in one division might get flagged in another. That uncertainty makes planning impossible.
What’s Changing in 2025?
The FDA knows this system is broken. That’s why they launched new programs to fix it.The ANDA Prioritization Pilot Program, started in September 2023, fast-tracks reviews for generics made in the U.S. with U.S.-sourced active ingredients. If you qualify, your PAS can be approved in as little as 8 months - down from the old 30-month average. The goal? Encourage companies to build manufacturing in America and reduce reliance on overseas suppliers.
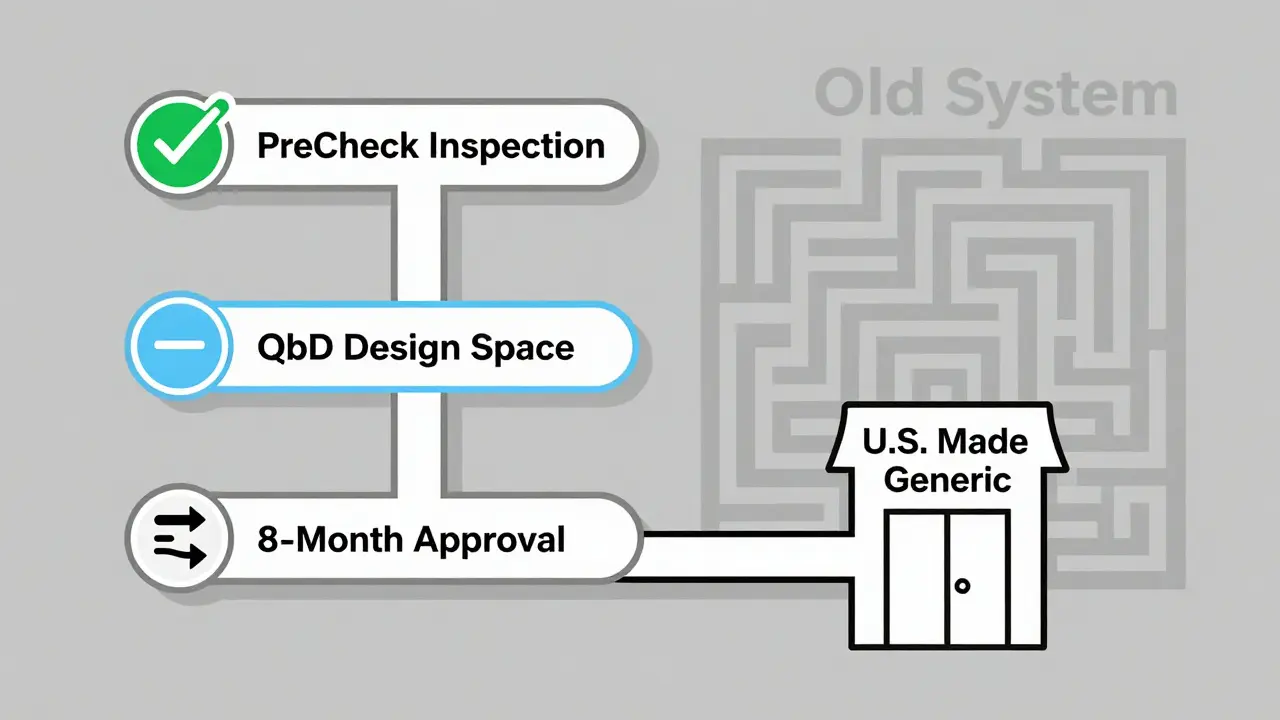
Then there’s the PreCheck program, launched in February 2024. It lets manufacturers get facility inspections done before they even submit a PAS. If your factory passes PreCheck, you can skip the long inspection wait during review. That cuts facility transfer times from 18 months to 9.
And in January 2024, the FDA released draft guidance proposing a new tiered system for complex generics. Under this model, minor changes to complex drugs might no longer need a full PAS - just a CBE or even an annual report. If finalized, this could reduce PAS submissions by up to 35%.
These changes aren’t just paperwork fixes. They’re economic incentives. The FDA estimates that by 2027, these programs could attract $4.2 billion in new U.S. manufacturing investment. That means more reliable supply chains, fewer drug shortages, and better access to generics.
How Can Manufacturers Reduce Re-Evaluation Risk?
The smartest companies don’t wait for a problem to trigger a PAS. They plan ahead.- Use Quality by Design (QbD): Build flexibility into the original ANDA. Define a “design space” - the range of conditions under which the product still performs the same. That way, future changes within that space don’t need approval.
- Invest in Process Analytical Technology (PAT): Real-time monitoring during production helps catch drift before it becomes a problem. Companies using PAT saw 32.6% fewer PAS submissions over five years.
- Do pre-submission meetings: Talk to the FDA before you submit. Ask: “Will this change need a PAS?” Their feedback can save months.
- Build a strong quality system: Companies with mature quality management systems cut approval times by over 30%.
Take Teva’s 2022 approval for continuous manufacturing of amlodipine. Instead of batch processing, they switched to a continuous line - a major change. But because they did extensive pre-submission meetings, shared real-time data, and used QbD principles, their PAS was approved in just 8 months. That’s faster than most CBEs.
What Should You Do If You’re a Patient?
You don’t need to understand the paperwork. But you should know this: if your generic drug suddenly looks different - color, shape, markings - it might be a new version. That’s normal. The FDA allows changes as long as the drug is still safe and effective. Your pharmacist should tell you if the formula changed. If you notice new side effects or the drug doesn’t seem to work as well, talk to your doctor. It’s not always the drug - but it’s worth checking.Bottom line: manufacturing changes are a necessary part of keeping generics safe and available. The system isn’t perfect, but it’s getting better. And with new FDA programs, the future looks less like a regulatory maze and more like a clear path forward.
What manufacturing changes require FDA approval before implementation?
Changes that significantly affect the drug’s safety, identity, strength, quality, or purity require a Prior Approval Supplement (PAS). These include switching manufacturing sites, changing the synthesis route of the active ingredient, scaling up production by more than 50%, introducing new excipients, altering the drug’s release profile, or modifying critical equipment. These changes must be approved by the FDA before you can implement them.
How long does a PAS submission take to get approved?
The average review time for a Prior Approval Supplement (PAS) is 10 months, but it can range from 8 to 18 months depending on complexity. Simple changes might be approved faster, while those involving new facilities, complex formulations, or missing data can take longer. The FDA may issue a complete response letter requesting more information, which adds time to the process.
Why do some generic manufacturers avoid making process improvements?
The cost and time of submitting a PAS - often $287,500 or more and taking over a year - can outweigh the benefits for low-margin generic drugs. Many companies fear unpredictable FDA feedback, extended delays, or rejection. As a result, they stick with outdated equipment or processes, even if they’re less efficient or more prone to failure, simply to avoid regulatory risk.
What is the ANDA Prioritization Pilot Program?
The ANDA Prioritization Pilot Program, launched in September 2023, fast-tracks FDA review for generic drugs manufactured and formulated in the U.S. using U.S.-sourced active ingredients. Qualifying applications can be approved in as little as 8 months - compared to the traditional 30-month average. The goal is to incentivize domestic manufacturing and reduce supply chain vulnerabilities.
Can a change in packaging trigger FDA re-evaluation?
Yes, if the packaging change affects the drug’s stability, protection from moisture, light, or oxygen, or alters how the drug is delivered (e.g., changing from blister packs to bottles). Even small changes like new labels or materials can require a PAS if they impact the drug’s shelf life or performance. The FDA requires stability data proving the drug remains within specifications under the new packaging.
How do Quality by Design (QbD) principles help reduce re-evaluation?
QbD helps manufacturers define a “design space” - the range of manufacturing parameters where the product still meets quality standards. If a change stays within this space, it doesn’t require a full PAS. By building flexibility into the original approval, companies can make future adjustments without triggering lengthy reviews, reducing PAS submissions by up to 40%.
Nancy Kou
This is such an important topic that nobody talks about. I work in pharma logistics and I’ve seen generics disappear overnight because a factory in India got a 30-day inspection delay. The FDA doesn’t care if patients are going without meds - they care about paperwork. It’s broken.
Hussien SLeiman
Let’s be real - the FDA isn’t protecting patients, they’re protecting Big Pharma’s monopoly. Every time a generic manufacturer tries to improve efficiency or cut costs, the FDA throws up a 14-month bureaucratic wall. Meanwhile, brand-name companies get to change their formulations with zero oversight. This isn’t safety - it’s market control disguised as regulation.
And don’t even get me started on how they treat U.S. manufacturers versus foreign ones. You think Teva got approved in 8 months because they’re better? No - they’re American. The system is rigged.
Why does a company need to prove that a new tablet press doesn’t alter bioavailability when the active ingredient hasn’t changed? Because the FDA doesn’t trust science - they trust forms. They’d rather wait 18 months for a 100-page PDF than trust a validated process.
And yet, we’re supposed to believe this system keeps us safe? I’ve had generic metformin from five different manufacturers. All worked the same. All looked different. None caused side effects. But according to the FDA, one of them might as well be poison because the binder was changed from cornstarch to potato starch.
It’s not about safety. It’s about control. And the cost? Billions in lost innovation. Thousands of jobs offshore. And patients paying more because no one dares to improve the process.
They call it ‘quality assurance.’ I call it ‘regulatory terrorism.’
Kevin Motta Top
Had a friend work at a generic lab in Indiana. They upgraded their coating machine - saved 20% on waste. Took 15 months to get PAS approved. During that time, they had to keep running the old machine that broke down every 3 days. Lost over $2M in downtime. They didn’t even get a thank you from the FDA.
Janelle Moore
Wait… so the FDA is letting foreign factories make our pills? And they’re not even checking them in person? I heard the FDA only inspects 1% of overseas plants. That’s not oversight - that’s negligence. What if they’re using Chinese talc that’s laced with asbestos? Or Indian labs that fake stability data? We’re all just guinea pigs. This is how people get cancer from generic blood pressure meds. They just don’t tell you.
And don’t believe them when they say ‘same medicine.’ The active ingredient might be the same, but the fillers? Those are chemicals. Some are linked to autism. Some cause liver damage. You think the FDA cares? They’re paid by the same lobbyists who own the brand-name companies. You’re being poisoned - slowly - and no one’s stopping it.
Henry Marcus
THEY’RE LYING TO YOU!! THE FDA DOESN’T TEST GENERIC DRUGS - THEY JUST READ PAPERS!! AND THE COMPANIES? THEY FUDGE THE NUMBERS!! I KNOW A GUY WHO WORKED AT A LAB IN CHINA - THEY WERE RECYCLING SOLVENTS AND CALLING IT ‘PURE’!! AND THE FDA? THEY’RE TOO BUSY TAKING LUNCH BREAKS TO NOTICE!!
EVERY TIME YOU TAKE A GENERIC, YOU’RE PLAYING RUSSIAN ROULETTE WITH YOUR LIVER!!
AND NOW THEY WANT TO SPEED UP APPROVALS?? ARE YOU OUT OF YOUR MIND?? WE NEED MORE INSPECTIONS - NOT FEWER!!
THEY’RE SELLING US OUT FOR PROFITS!!
William Liu
This is actually really hopeful. The new programs are a big step forward. More U.S. manufacturing means more jobs, fewer shortages, and better quality control. It’s not perfect, but it’s moving in the right direction. We need to support companies that are trying to do the right thing - even if it’s slow and expensive.
Andrew Kelly
Let me be clear - the FDA is not an enemy. They’re a necessary evil. But the system is absurd. A company spends $300K to change a filter and gets a letter back asking for a third-party validation of the cleaning procedure for a machine that hasn’t been touched in 12 years. This isn’t science - it’s medieval bureaucracy.
And yet, I’ve seen companies that play the game well. They hire ex-FDA reviewers as consultants. They pre-submit. They use QbD. They don’t fight the system - they weaponize it. The ones who complain the most? They’re the ones who didn’t plan.
It’s not about being anti-FDA. It’s about being pro-efficiency. And if we want generics to stay affordable, we need to fix this - not vilify it.
Isabel Rábago
People don’t understand - this isn’t about money. It’s about ethics. If you change how a drug is made, you have a moral obligation to prove it’s still safe. Not because the FDA demands it - because patients deserve it. I’ve seen kids on epilepsy meds whose seizures returned after a generic switch. Not because the drug was bad - because the dissolution rate changed by 7%. That’s all it took.
So yes, it’s slow. Yes, it’s expensive. But if you’re cutting corners on medicine, you’re not saving money - you’re stealing health.
Dev Sawner
It is imperative to note that the regulatory framework governing generic pharmaceuticals is not merely a procedural formality, but a critical safeguard for public health. The stringent requirements for Prior Approval Supplements ensure that bioequivalence is not compromised, regardless of geographical or operational variables. The current review timelines, while seemingly protracted, are commensurate with the complexity of pharmaceutical manufacturing and the non-negotiable imperative of patient safety. Any attempt to expedite the process without robust analytical validation constitutes a grave risk to the integrity of the healthcare ecosystem.
Moses Odumbe
Bro, the FDA is like your ex who still texts you at 2 a.m. saying ‘did you take out the trash?’ - but you already did it 3 times. They need to chill. I’ve seen labs use AI to predict dissolution profiles - and the FDA still wants 500 pages of manual HPLC reports. We’re in 2025. Use machine learning. Stop asking for paper.
Also, emoji for the win 🤖💊
Meenakshi Jaiswal
If you’re a small generic manufacturer, don’t panic. Start small. Do a pre-submission meeting. Use QbD even if it’s just for one excipient. Talk to the FDA like a partner, not a cop. I helped a startup in Pune get their PAS approved in 9 months - they just prepared like they were presenting to investors. Clear data. Clear goals. No fluff. It’s possible.
You don’t need to be Teva. You just need to be organized.
bhushan telavane
India makes 40% of the world’s generics. We’ve been doing this for decades. The FDA’s rules are fine - but they need to understand that not every change is a threat. Sometimes, it’s just better packaging. Or a cheaper supplier. We’re not trying to fool anyone. We’re trying to feed the world.
Kelly Mulder
So let me get this straight - a company can change the shape of a pill, but if they use a different dye, it’s a 14-month nightmare? And you’re telling me this isn’t about controlling the market? The FDA is a corporate puppet. They don’t care about patients. They care about lawsuits. And if you’re a small company? You’re dead in the water. This isn’t regulation. It’s extinction.
Takeysha Turnquest
What is safety but a story we tell ourselves to sleep at night? The pill looks the same. The label says the same. But the soul of the medicine - the rhythm of its release, the whisper of its impurities - that’s changed. And no one is listening. The FDA doesn’t hear the silence between the molecules. Only the noise of the forms.