
When we talk about Cystic Fibrosis is a life-limiting autosomal recessive genetic disorder caused by mutations in the CFTR gene, we're talking about a fundamental glitch in how the body handles salt and water. This isn't a contagious illness you catch; it's written into the DNA. To have the condition, a person must inherit two mutated copies of the gene-one from each parent. If you only have one, you're a carrier. You won't feel sick, but you can pass that gene to your children. It's a hidden blueprint that, when doubled, changes the very chemistry of the body's secretions.
The Biology of Sticky Mucus
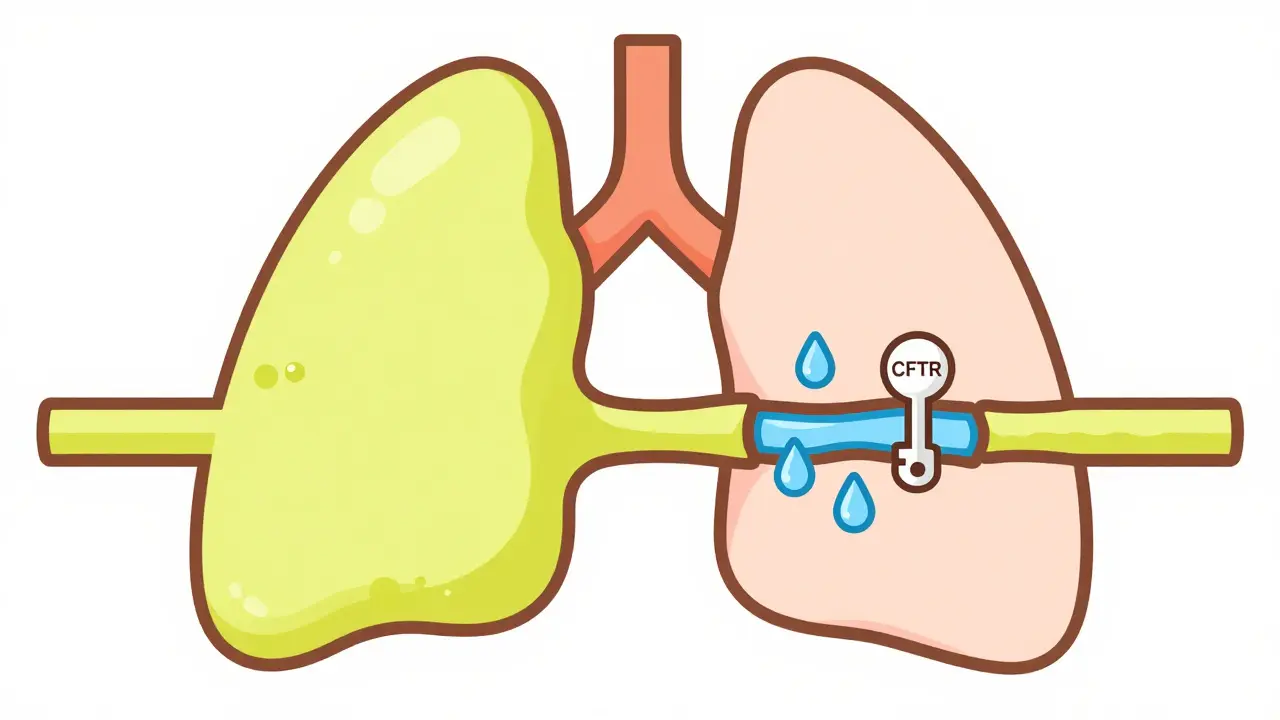
At the heart of the struggle is the CFTR Protein (Cystic Fibrosis Transmembrane Conductance Regulator). Think of this protein as a tiny gateway on the surface of cells that lets chloride ions move in and out. When this gateway is broken or missing, the balance of salt and water on the cell surface collapses. The result? Mucus that should be thin and slippery becomes thick, sticky, and incredibly stubborn.
This "mucus trap" creates a domino effect across the body. In the lungs, the thick slime blocks the airways, turning the respiratory system into a breeding ground for bacteria like Pseudomonas aeruginosa. Because the mucus won't move, the body triggers a constant state of inflammation, which slowly scars the lung tissue. This is why respiratory failure is the leading cause of death for about 85% of patients. But the damage isn't limited to the chest. The pancreas also gets clogged, preventing essential enzymes from reaching the gut, which leads to malnutrition and poor growth in children.
| Organ System | What Happens | Key Impact |
|---|---|---|
| Lungs | Mucus plugs the bronchial tubes | Chronic infections and scarring |
| Pancreas | Blocked digestive enzyme ducts | Malnutrition and insulin issues |
| Liver | Bile duct obstruction | Biliary cirrhosis in ~30% of cases |
| Reproductive | Absence of vas deferens (males) | Infertility in 97-98% of men |
The Genetic Blueprint: Why Every Case is Different
Not all cases of CF are the same. There are over 2,000 known mutations of the CFTR gene located on chromosome 7. The most frequent culprit is the F508del mutation, which is found in roughly 70% of people globally. Depending on which mutation you have, the protein might be completely missing, might not fold correctly, or might just be "leaky."
This genetic diversity is why the medical community moved toward precision medicine. For years, we treated the symptoms-coughing up mucus and taking enzymes. Now, we treat the protein itself. This shift has been a game-changer. In the 1960s, the median survival age was a heartbreaking 14 years. Today, thanks to targeted therapies, that number has climbed to nearly 51 years. That is a massive victory for science and the patients who fought for it.
The Era of CFTR Modulators
The real breakthrough came with CFTR Modulators. Unlike traditional medicine, these drugs don't just clear the mucus; they fix the broken gateway. Depending on the mutation, modulators either help the protein reach the cell surface (correctors) or help the gate open wider (potentiators).
The most advanced version is triple combination therapy, known commercially as Trikafta. By combining three different drugs-elexacaftor, tezacaftor, and ivacaftor-it targets a wider range of mutations, including the common F508del. The results are often immediate and dramatic. Some patients have reported their daily airway clearance time dropping from 90 minutes down to just 20 minutes. Imagine getting an hour of your life back every single day.
However, these miracle drugs aren't a perfect cure for everyone. About 10% of the CF population has "nonsense mutations" that current modulators can't touch. For these individuals, the gap in care is a critical issue. Furthermore, the cost is staggering-often reaching $300,000 per year in the US-making these life-saving drugs a luxury that many in low-income countries simply cannot access.

Managing the Daily Grind
Even with new drugs, living with CF is a full-time job. For those not on modulators, the daily treatment burden can take 2 to 3 hours. It's a rigorous routine that requires immense discipline. A typical day involves:
- Airway Clearance: Using chest physiotherapy or oscillating devices to shake the mucus loose from the lungs.
- Inhaled Medications: Using 4 to 6 different nebulized medications to thin the mucus and fight infections.
- Enzyme Replacement: Taking 6 to 12 capsules of pancreatic enzymes with every single meal to absorb nutrients.
- High-Calorie Diet: Eating significantly more than the average person to combat malabsorption.
The learning curve is steep. Most patients need several supervised sessions with a respiratory therapist just to get the airway clearance techniques right. It's a grueling cycle, but the goal is simple: keep the lungs clear and the body nourished to avoid the dreaded "pulmonary exacerbation"-a sudden worsening of symptoms that often requires hospitalization.
The Horizon: Gene Editing and Beyond
We are moving toward a future where we don't just "modulate" the protein, but actually fix the DNA. Researchers are currently exploring mRNA Therapy and gene editing tools like CRISPR. The goal is to deliver a healthy copy of the CFTR gene directly into the lung cells.
There is also a push to develop better anti-infectives. Because bacteria like Pseudomonas become resistant to drugs when trapped in thick mucus, scientists are testing liposomal ciprofloxacin, which delivers the antibiotic more effectively into the biofilm. While we aren't at a total cure yet, the trajectory is clear. We've moved from managing a terminal childhood illness to optimizing adult life, and the next step is erasing the genetic error entirely.
What is the gold standard test for diagnosing Cystic Fibrosis?
The sweat chloride test is the diagnostic gold standard. Because CF affects salt transport, people with the condition have abnormally high levels of chloride in their sweat. Typically, a sweat chloride level greater than 60 mmol/L is indicative of Cystic Fibrosis.
Can people with CF have children?
Yes, though it is more complex for men. About 97-98% of men with CF are infertile because they are born without the vas deferens (the tube that carries sperm). However, sperm can often be retrieved surgically for IVF. Women with CF can conceive, although thick cervical mucus can sometimes make natural conception more difficult.
Are there side effects to CFTR modulator therapies?
While generally well-tolerated, some patients experience side effects. A small percentage (around 3.2%) have reported severe elevations in liver enzymes, which may require them to stop the medication. Regular liver function monitoring is usually required during treatment.
How does CF differ from Primary Ciliary Dyskinesia (PCD)?
Both cause respiratory issues, but the cause is different. PCD is a problem with the "brushes" (cilia) that sweep mucus out of the lungs. CF is a problem with the "fluid" (mucus) itself, which is too thick to be swept away. Also, CF affects the pancreas and liver, whereas PCD is primarily respiratory and sinus-related.
When is CF typically diagnosed?
In most developed regions, CF is now detected shortly after birth through newborn screening programs. This allows for early intervention with nutrition and respiratory therapy, which significantly improves long-term lung health.
Betty Kawira
The daily routine for CF is absolutely brutal, but having a high-quality percussion vest or a newer Acapella device makes a world of difference in getting that junk out of the lungs.
Anyone still doing manual chest physiotherapy should really look into the newer oscillating PEP devices to save some time.
prince king
It is truly humbling to see how science can rewrite a destiny that once seemed set in stone! 🧬✨ The progress from childhood tragedy to adult longevity is a beautiful testament to human ingenuity and persistence. Keep fighting the good fight everyone! ❤️🌟
Peter Minto
300k a year is absolute robbery! We got the best meds in the US and some greedy pharma suit is just robing us blind. Its a discrace to the flag that we can't get this sorted out without paying a ransom for a pill!!
Darrin Oneto
That mucus trap sounds like a real nightmare fuel scenario. Glad the new gear is kickin' some butt and givin' folks their lives back. Totally wild how a tiny glitch in the code messes up the whole machine, huh?
Dale Kensok
The reductionist approach to CFTR modulation, while empirically successful, barely scratches the surface of the epigenetic complexities involved. One must consider the stochastic nature of protein folding and the systemic homeostasis disrupted by such targeted interventions.
The mere quantification of 'minutes saved' in airway clearance is a pedestrian metric that fails to capture the qualitative shift in the patient's ontological experience of their own pathology. We are merely treating the phenotype while the genotype remains an immutable, haunting presence. This is the tragedy of the modern medical industrial complex: providing a pharmacological veil over a structural biological failure. The pursuit of CRISPR is the only intellectually honest path toward resolution, though the ethical ramifications of germline editing remain a quagmire of deontological contradictions. Until we transcend the rudimentary modulator phase, we are simply refining the art of the bandage.
Timothy Brown
Honestly, people who complain about the 'grind' of the treatment just aren't disciplined enough. If you actually care about your health, you make the time. It's not that hard if you just optimize your schedule.
Jean Robert
I just want everyone to know that it is completely okay to feel overwhelmed by the weight of a lifelong diagnosis, and it's important to remember that you aren't alone in this journey because there are so many wonderful communities out there ready to hold your hand and cheer you on through every single nebulizer treatment and every difficult hospital stay. Please be gentle with yourselves because you are doing the best you can with a very difficult hand, and every single breath you fight for is a victory worth celebrating with all the love in the world.
Amber McCallum
Life is all about balance. If you have a glitch in your DNA, maybe your soul is just trying to tell you something about your spiritual path. Just accept it.
Jarrett Jensen
The provided exposition on the F508del mutation is rudimentary at best. It is quite tiresome to see such simplistic summaries when the molecular dynamics are far more nuanced.
Steve Grayson
The point about the 10% with nonsense mutations is really important. We can't leave them behind while the rest get better.
Trish Perry
It makes me think about how we define 'normal' when our very chemistry is different. The struggle with the mucus is physical, but the mental load of being a 'full-time patient' is where the real battle happens.
Thomas Jorquez
The science is quite impressiv, though the cost remains a significant barrier for many. It is a complex situation for the healthcare system.
Justin Crice
The efficacy of liposomal ciprofloxacin in penetrating the Pseudomonas biofilm is a fascinating area of pharmacokinetics. The altered lipid bilayer allows for superior intracellular delivery compared to standard aqueous formulations.
Michael Yoste
I totally feel you on the struggle! It's so hard when the world doesn't get it, but we're all in this together, right? Just lean into the love!